Overview
We have expanded our representation of gnomAD v4 data to include data specifications from the Genomic Knowledge Standards Work Stream (GKS) of the Global Alliance for Genomics & Health (GA4GH). This work has been done as part of a national initiative to Improve the AI/ML-Readiness of NIH-Supported Data (NOT-OD-22-067).
This release includes:
- Addition of Variation Representation Specification (VRS 1.3) objects to the gnomAD v4 exome and genome Hail Tables
- Implementation of a Cohort Allele Frequency model for use in the Variation Annotation (VA) framework
- Access methods for GKS-formatted data within the gnomAD Hail Utilities
These GA4GH specifications provide a framework for computable genomic evidence and knowledge structures. The framework enables interoperable representation of evidence for scalable, federated variant interpretation in genomics research, clinical genetic testing, and clinical decision support systems.
Introduction
The Genome Aggregation Database is a key dataset used in the clinical interpretation of patient genomes. Under existing frameworks, such as the ACMG/AMP guidelines for pathogenicity1 and the ClinGen/CGC/VICC oncogenicity guidelines2, allele frequency evidence can be highly impactful for determining whether an observed variant is likely to contribute to an observed clinical condition. Often, the presence of high frequency variants in gnomAD is sufficient standalone evidence to classify a variant as benign for rare Mendelian disease. While some databases such the ClinGen Evidence Repository3 and CIViC4 codify how knowledge from gnomAD and other sources is applied under clinical classification frameworks, AI/ML readiness of these data can be improved by adopting semantic standards for representing the underlying evidence (e.g. gnomAD allele frequency data) used to determine what evidence classification codes are used (Figure 1).
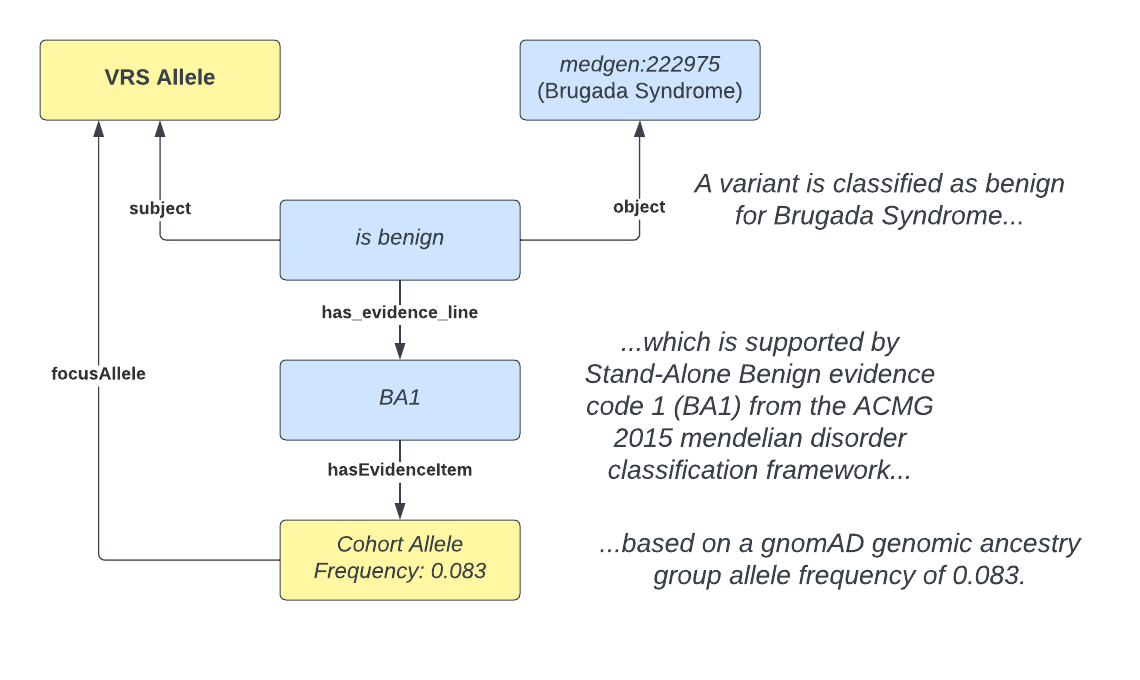
Standards that work interchangeably across known and novel classes of genomic evidence are a necessary component for realizing an AI/ML-ready genomic knowledge architecture. In 2019, the GA4GH approved the Variation Representation Specification (VRS) as a standard for the computational representation and exchange of genomic variation data as the founding specification for this genomic knowledge framework. VRS5 is a terminology, information model, schema, and conventions for the computational representation of genomic variation. These features enable federated, globally unique identification of variants for the precise exchange of genomic variation data between computer systems. Since its initial release, VRS has been extended to precisely define and disambiguate multiple forms of variation, including alleles, haplotypes, copy number variants, and genotypes6. The VRS reference implementation, VRS Python, is readily installed via the Python Package Index (PyPI), and source code is publicly available on GitHub. Educational notebooks within the repository provide examples of different VRS functionalities and can be run locally or on the cloud on Binder or on Terra.
Along with VRS, the GA4GH is developing a modeling framework and machine-readable schema to represent genomic knowledge statements, study results, and related information associated with genomic variants. Described as the Variation Annotation specification (VA-Spec), this framework is founded on the principles of the Scientific Evidence and Provenance Information Ontology (SEPIO)7 and is designed to formalize the structure and semantics of genomic evidence to computationally derive conclusions regarding variation in a federated genomic knowledge ecosystem.
Sources such as gnomAD report cohort allele frequency (CAF) evidence under the VA-Spec framework. CAF results describe the frequency of an Allele among a group of people defined by one or more shared characteristics (a cohort). In gnomAD, cohorts are known as genetic ancestry groups, where genetic ancestry is the definitional property for defining sub-cohorts from which the allele frequency calculations are derived. In other resources, cohort frequency data may be defined by characteristics such as sex, phenotype, or disease. With the v4 release, gnomAD is pioneering the cohort allele frequency statement type, paving the way for the use of gnomAD in federated, AI-driven research and clinical applications.
Technical Details
One of the defining features of VRS is the ability to create computed identifiers for genomic variation that are globally consistent and unique5. Using the VCF annotation tool from VRS-Python v0.8.4, we annotated the v4 Hail Tables with VRS variant identifiers and associated data for constructing complete VRS data objects. This work required computation of Alleles using VRS conventions, requiring the rapid sequence retrieval and VOCA normalization methods5,8 supported by the Biocommons seqrepo9 and bioutils packages, respectively (Figure 2). We annotated 183.6 million variants called from the gnomAD v4 exomes, 759.3 million variants called from the gnomAD v4 genomes, and an equivalent number of reference-match alleles, for a total of ~1.89 billion variants.
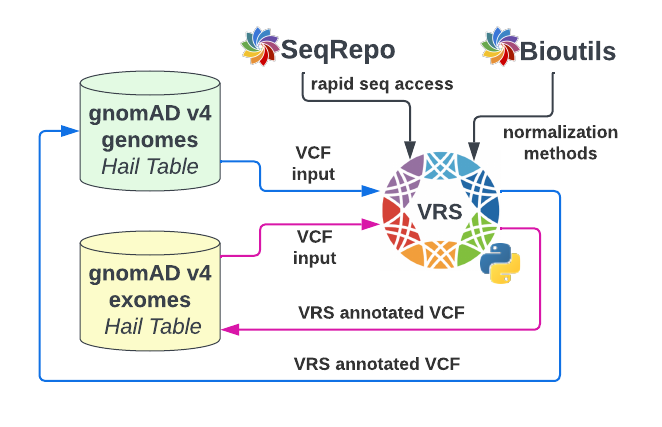 * Figure 2 - VRS annotation pipeline with VRS Python and Biocommons. Data from the gnomAD v4 genome and exome hail tables were individually annotated with the VRS-Python VCF annotation tool through intermediate VCF files. High-throughput sequence retrieval and variant normalization operations in VRS Python are supported by the Biocommons SeqRepo and Bioutils packages, respectively.*
* Figure 2 - VRS annotation pipeline with VRS Python and Biocommons. Data from the gnomAD v4 genome and exome hail tables were individually annotated with the VRS-Python VCF annotation tool through intermediate VCF files. High-throughput sequence retrieval and variant normalization operations in VRS Python are supported by the Biocommons SeqRepo and Bioutils packages, respectively.*
Accessing the VRS records from the gnomAD Hail Tables is supported by the Hail utilities for gnomAD, an open-source python library maintained by the gnomAD team. The VRS variant representation for each gnomAD allele is embedded in the CAF object (Figure 3) returned by the gnomad_gks() method (documentation). The CAF object can optionally include or omit CAF data for individual genetic ancestry groups, include further subdivision of genetic ancestry groups by sex, or return only the associated VRS object (without allele frequency information). In addition, this method may retrieve CAF data from a custom Hail Table in lieu of the gnomAD public data release. Similarly, users have the option to include mean coverage information, either from the gnomAD public release coverage Hail Table table or from a custom Hail Table containing coverage information.
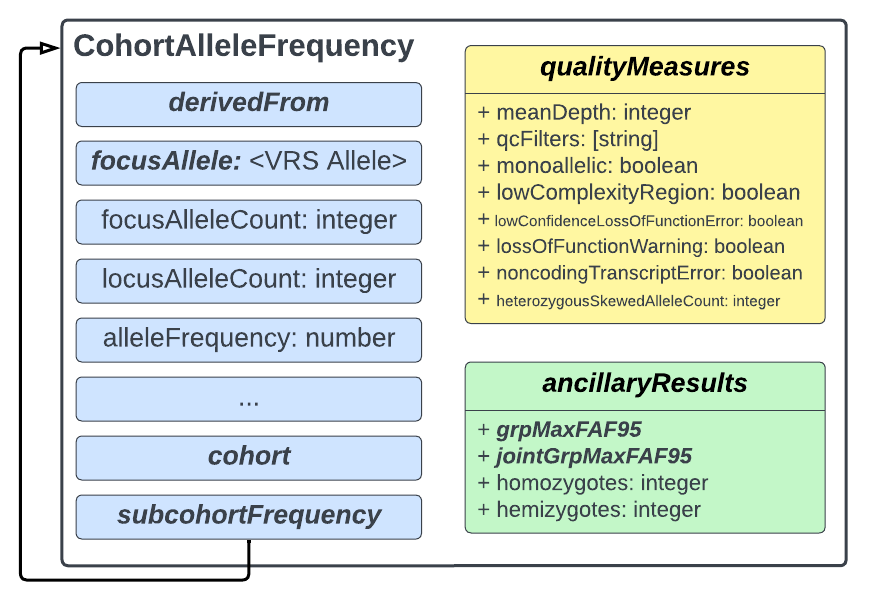
While much of the structure of the CAF model reflects similarly named fields in the gnomAD resource (Figure 3), there are a few key terminological differences. As mentioned in the introduction, the CAF model refers to people groups as cohorts, and divisions of the cohort as subcohorts. This allows the model to be used across gnomAD and other resources that may define allele frequency on cohorts divided by characteristics other than genetic ancestry. CAF statements also have a focus allele, which is the VRS representation of the variant described by a gnomAD record. The CAF model thus describes the integer count of these alleles in the cohort as the focus allele count, which is similar to the use of “Allele Count (AC)” in gnomAD. In contrast, where the gnomAD data model uses an “Allele Number (AN)”, the equivalent concept in the CAF model is the locus allele count, as this represents a count of all Alleles (ref and alt) observed at the genomic locus. New in gnomAD v4 is the 95% confidence interval joint group max filtering allele frequency, which is captured by the jointGrpMaxFAF95 attribute. This attribute captures details about the calculated maximum population frequency called jointly across the gnomAD v4 exome and genome datasets. This field accompanies the similar exome- or genome-specific calculation transmitted in the grpMaxFAF95 attribute.
Conclusion
Under the auspices of the GA4GH GKS Work Stream, we have developed and deployed a draft, interoperable model for the representation of allele frequency data from genetic ancestry groups. The new cohort allele frequency (CAF) model resulting from this effort balances generalized information about allele frequency data from person groups (cohorts), with resource-specific features unique to gnomAD (e.g. joint group max filtering allele frequency). In performing this work, we have demonstrated the feasibility of VRS annotation and retrieval on very large genomic datasets, and developed open-source reference methods for CAF evidence retrieval through the Hail utilities for gnomAD.
Despite these advancements, we continue to work to improve the representation of gnomAD v4 data under the auspices of the GA4GH GKS Work Stream. Specifically, while the CAF model is part of the draft VA-Spec, it is at an early maturity stage and the model may still change during the VA-Spec product development and approval cycle. In addition, work on the next major version of VRS (v2.0) is under development and will include important new features, including structural variant (SV) representation, built-in variant decoration, and URI support. We anticipate future annotations to the gnomAD v4 Hail tables to accommodate the v2 data model and associated computed identifiers. Finally, we are actively working to add support for the CAF model in the gnomAD GraphQL API.
The addition of the GA4GH VRS and CAF specifications in gnomAD is a key step towards federated, interoperable genomic evidence. By providing access to the gnomAD v4 data in this format, we are realizing our objectives to maximize reuse of the gnomAD data across interpretation platforms of today, while laying the groundwork for the AI-based variant interpretation tools of tomorrow. Through collaboration with the GA4GH, gnomAD v4 is leading the way towards a more interoperable future.
Acknowledgments and Contact Information
We would like to acknowledge the National Human Genome Research Institute (award R35HG011949-02S1) for supporting development of this feature. Questions about the work described in this post may be directed to Alex.Wagner@nationwidechildrens.org. Feature requests for GA4GH VRS, VA-Spec, and the Hail utilities for gnomAD may be submitted at their linked issue trackers, respectively.
References
- Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
- Horak, P., Griffith, M., Danos, A. M., Pitel, B. A. & Madhavan, S. Standards for the classification of pathogenicity of somatic variants in cancer (oncogenicity): Joint recommendations of Clinical Genome Resource (ClinGen) …. Genet. Med. (2022).
- Preston, C. G. et al. ClinGen Variant Curation Interface: a variant classification platform for the application of evidence criteria from ACMG/AMP guidelines. Genome Med. 14, 6 (2022).
- Krysiak, K. et al. CIViCdb 2022: evolution of an open-access cancer variant interpretation knowledgebase. Nucleic Acids Res. 51, D1230–D1241 (2023).
- Wagner, A. H. et al. The GA4GH Variation Representation Specification: A computational framework for variation representation and federated identification. Cell Genom 1, (2021).
- Goar, W. et al. Development and application of a computable genotype model in the GA4GH Variation Representation Specification. Pac. Symp. Biocomput. 28, 383–394 (2023).
- Brush, M. H., Shefchek, K. & Haendel, M. SEPIO: A semantic model for the integration and analysis of scientific evidence.
- Holmes, J. B., Moyer, E., Phan, L., Maglott, D. & Kattman, B. SPDI: data model for variants and applications at NCBI. Bioinformatics 36, 1902–1907 (2020).
- Hart, R. K. & Prlić, A. SeqRepo: A system for managing local collections of biological sequences. PLoS One 15, e0239883 (2020).