Today we are pleased to announce the incorporation of cumulative counts of gnomAD individuals carrying pairs of rare co-occurring variants within genes in the gnomAD v2 browser, across various allele frequencies and functional consequences. These counts can be used to evaluate how frequently rare variant co-occurrence is observed in a large reference population. We envision that this data will aid the medical genetics community in interpreting the clinical significance of rare co-occurring variants found in patients, in the context of autosomal recessive disease. This feature builds off of our variant co-occurrence (inferred phasing) work (see “Variant Co-Occurrence (Phasing) Information in gnomAD”).
Background
A pair of variants in a gene can occur in cis (same copy of the gene) or trans (different copies of the gene), and the variants can be damaging to gene function (e.g., loss-of-function) or can be neutral. These distinctions have important consequences for interpreting the potential of a variant pair to cause an autosomal recessive disease, whereby both copies of a gene must carry a variant (i.e., occur in trans) and the variants must be damaging.
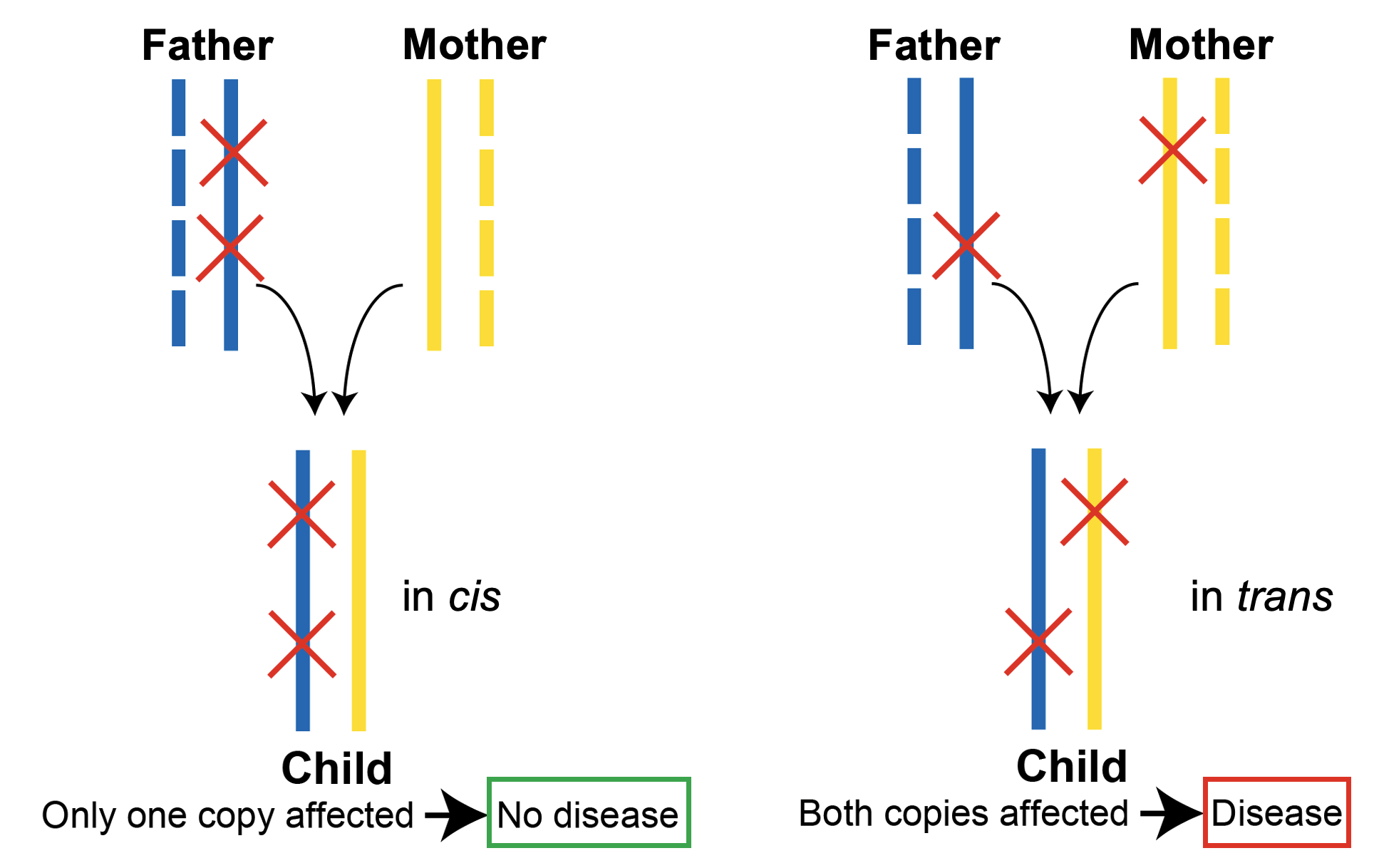
In cis and in trans variant pairs are often indistinguishable when using short read sequencing data. In the vast majority of scenarios, the two variants are too far apart to be captured on the same short read. However, with the release of variant co-occurrence (inferred phasing) information for gnomAD v2 exomes, it is now possible to infer phase with high confidence. This presents the opportunity to determine whether individuals in gnomAD carry compound heterozygous (in trans) rare damaging variants in a gene—a scenario that would not be expected for genes implicated in autosomal recessive conditions in a reference population depleted for severe Mendelian disease.
Approach
For each gene, we count the number of individuals carrying pairs of rare variants by inferred phase, allele frequency, and predicted functional consequence.
We inferred phase (in trans, unphased, or in cis) using the probability of two variants being in trans (Ptrans) (see “Variant Co-Occurrence (Phasing) Information in gnomAD”). We annotated variant pairs with Ptrans ≥ 0.55 as in trans, and variant pairs with a Ptrans ≤ 0.1 as in cis. We labeled as unphased all singleton-singleton variant pairs (i.e., variant pairs where both variants in the pair are only seen in one individual in gnomAD) as well as all variant pairs with indeterminate phase predictions (0.10 < Ptrans < 0.55).
Next, we set thresholds for the popmax1 allele frequency and global allele frequency, ranging from ≤ 0.5% to ≤ 5%. We annotated variant pairs with the highest popmax or global allele frequency of the variants in the pair and excluded variant pairs containing a variant with an allele frequency > 5% in any population that is not included in popmax.
We annotated each variant with the worst functional consequence on the canonical transcript (e.g., loss-of-function > missense > synonymous). We only annotated high-confidence (as predicted by LOFTEE) predicted loss-of-function (pLoF) variants in order to increase our confidence in predicted damaging variant consequences. In line with ClinGen recommendations for the interpretation of REVEL scores, we annotated missense variants as having “strong” (REVEL ≥ 0.932), “moderate” (REVEL ≥ 0.773), or “weak” (REVEL ≥ 0.644) levels of evidence for variant pathogenicity. Missense variants below the “weak” REVEL threshold were simply annotated as “missense”.
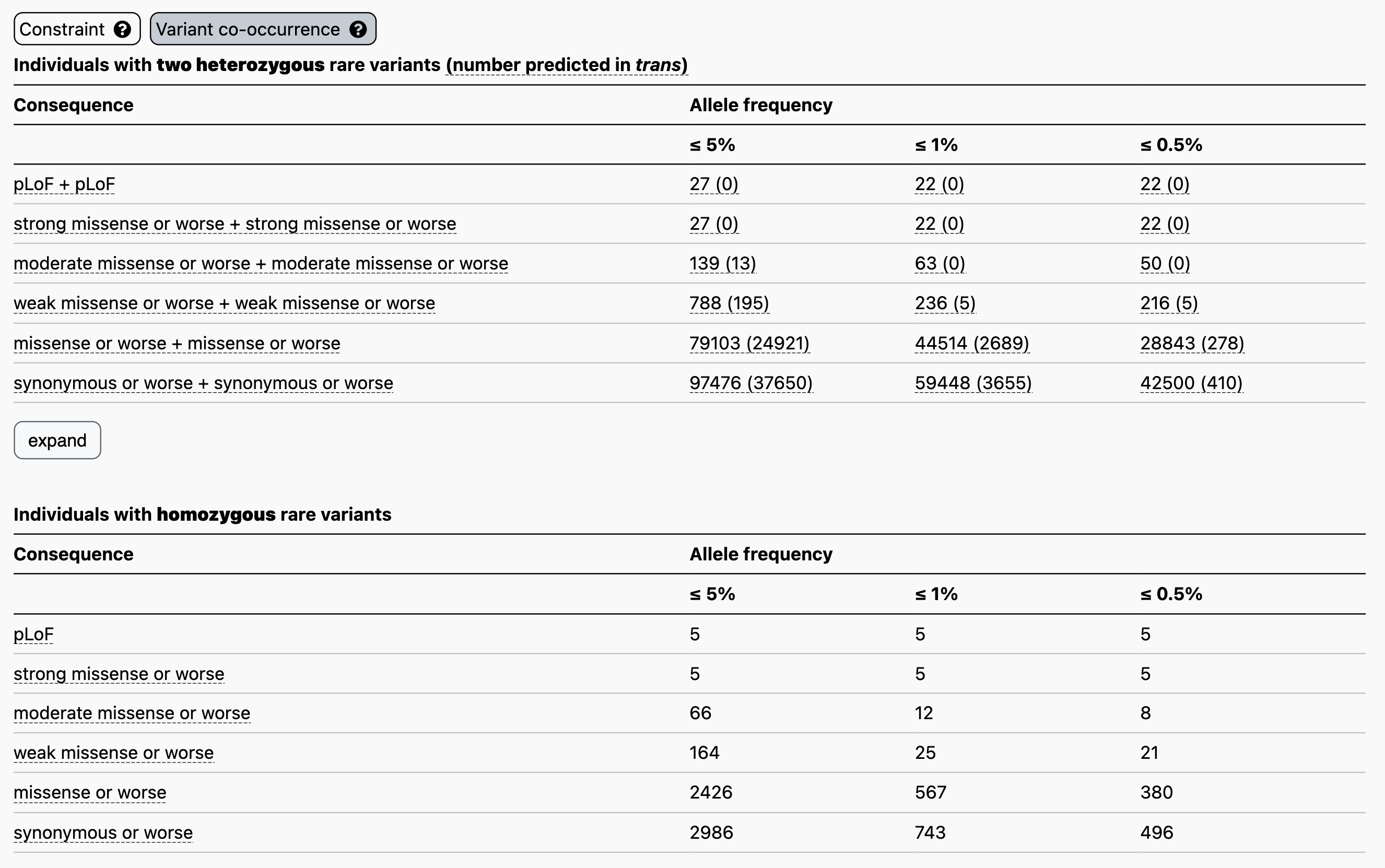
For comparison, we tabulated the number of individuals with homozygous variants in the same manner across pLoF variants (as previously reported), various tiers of evidence strength for predicted pathogenicity for missense variants, and synonymous variants.
As depicted below, we found only a small number of genes carrying rare predicted in trans damaging variants: 32 genes with pLoF and/or strong missense variants at ≤ 1% allele frequency, in comparison to a substantially higher number in cis (1,922 genes). The majority of genes with variants in trans (26/32 genes) are not disease associated in OMIM, in keeping with our expectation for a reference population depleted of severe Mendelian disease.
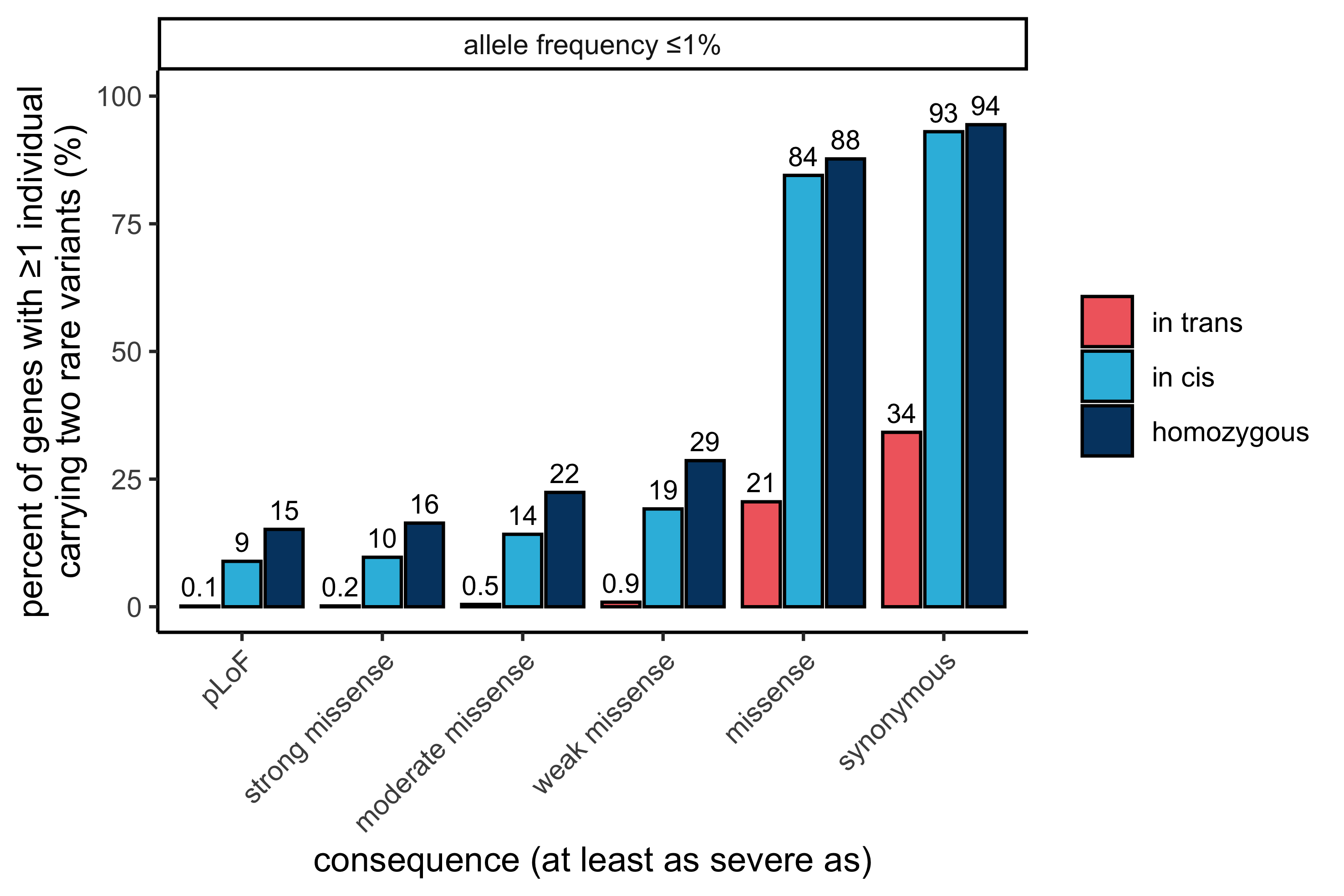
In this display, both variants in the variant pair must be annotated with a consequence at least as severe as the consequence listed (i.e., a variant pair consisting of a pLoF variant and a strong missense variant is not counted in “pLoF” but is counted in “strong missense” and all other displayed less severe consequences).
How to use the browser
On the top right hand corner of the gene landing page, users can select the variant co-occurrence tab (next to the constraint tab) to display a table with the counts of individuals carrying two rare variants across the gnomAD v2 exomes (n=125,748). In the main table, allele frequency thresholds of ≤ 5%, ≤ 1%, and ≤ 0.5% are displayed across six predicted functional consequences (combinations of pLoF, various evidence strengths of predicted pathogenicity for missense variants, and synonymous variants). In all cases, both variants in the variant pair must be annotated with a consequence at least as severe as the consequence listed (i.e., pLoF + moderate missense also includes pLoF + strong missense and pLoF + pLoF).
Importantly, individuals are counted once within each category. Therefore, if an individual carries more than two rare variants in a gene (e.g., three moderate missense variants), they will only be counted once despite multiple pairwise combinations. For display in the browser, we prioritize counting variant pairs with evidence of being in trans over unphased variant pairs, and unphased variant pairs over variant pairs with evidence of being in cis.
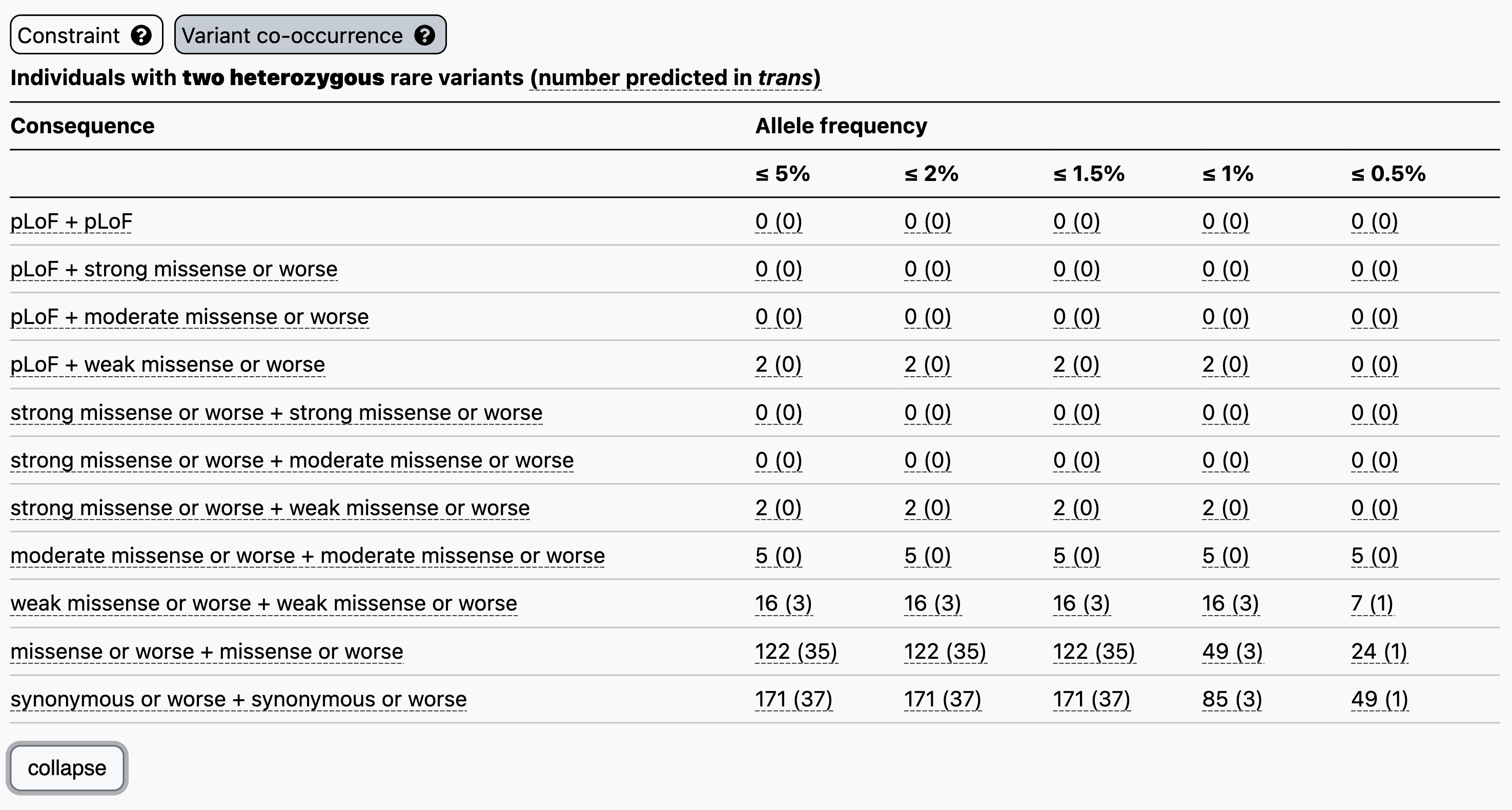
Let’s use the recessive disease gene GBA1 as an example. When searching for GBA1 on the main gnomAD landing page and clicking on the “Variant co-occurrence” tab on the right, you will see the display below. Two counts tables are displayed broken down by allele frequency and functional consequence, one for two heterozygous rare variants (above) and one for homozygous variants (below).
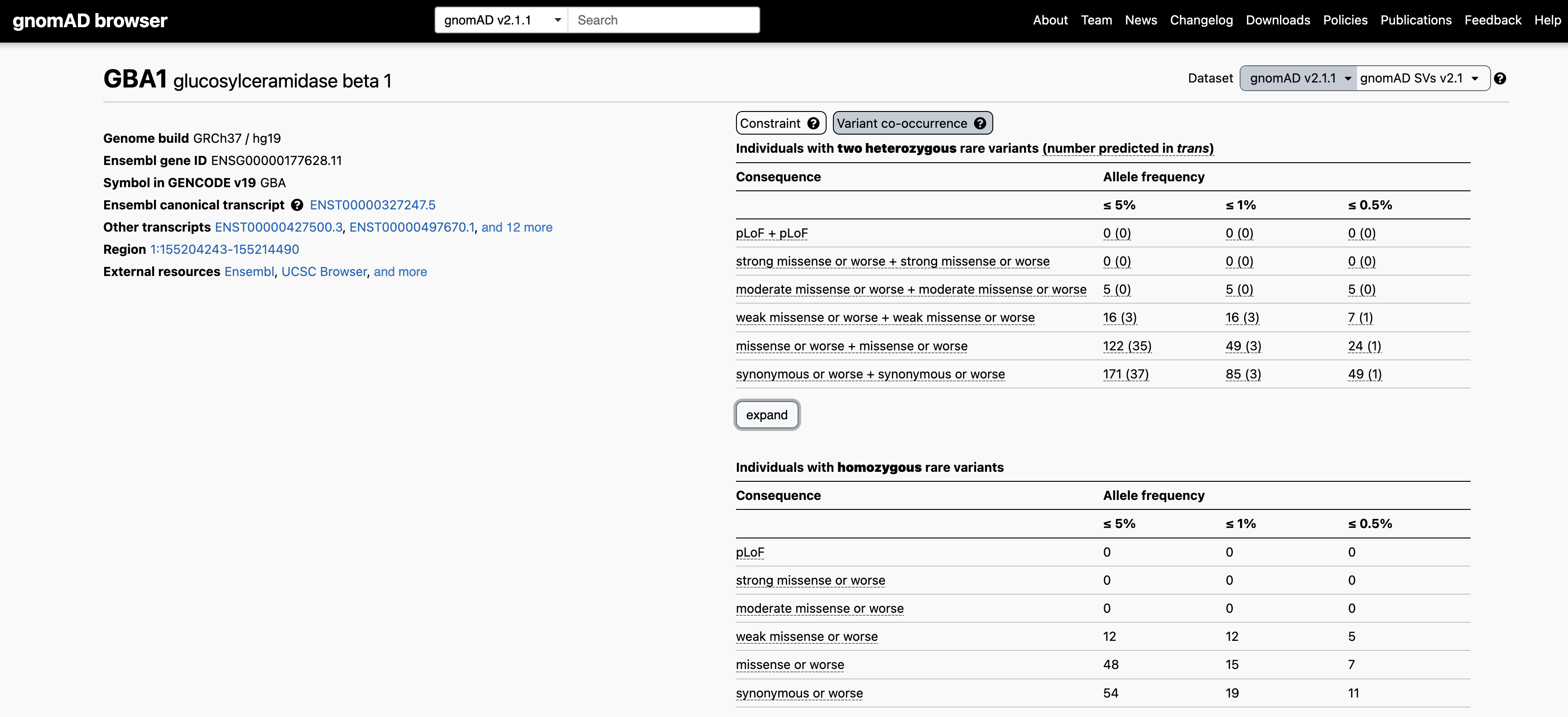
Focusing on the two heterozygous rare variants table, for each gene we list the number of individuals carrying any two variants, irrespective of phase, followed in parentheses by the number of individuals carrying two variants with evidence of being in trans. Hovering over these counts will also display the number of individuals carrying variants that are unphased and the number that have evidence of being in cis.
These counts tables can be used to determine the probability of observing a specific rare variant co-occurrence situation in the reference population and can aid in interpreting variant combinations found in patients with a suspected autosomal recessive Mendelian disease.
For example, let’s say you have a proband with two rare heterozygous missense variants with REVEL scores in the moderate range in GBA1. The counts table demonstrates that, at ≤ 5% allele frequency, 5/125,748 individuals in gnomAD carry two variants of this type, all of which have evidence of being in cis. Overall, low levels of co-occurrence in the reference population indicate low probability that your observation in the proband has occurred by chance.
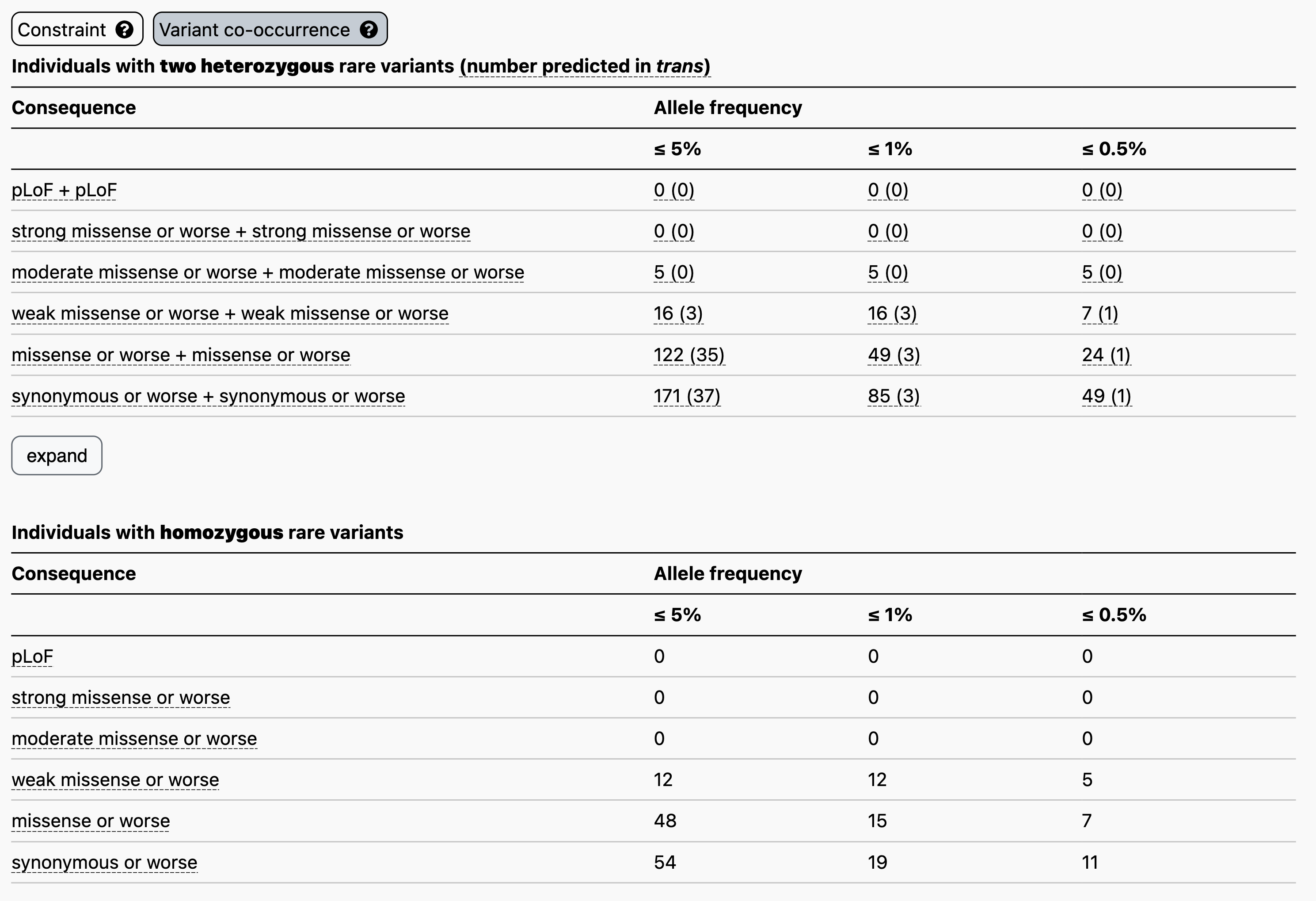
Clicking “expand” just below the table, will display additional allele frequency thresholds (5%, 2%, 1.5%, 1%, and 0.5%) and additional combinations of functional consequences.
Now let’s say you have a proband with two rare heterozygous missense variants in the disease gene TTN. Here, at ≤ 1% allele frequency, the counts table demonstrates that 44,514/125,748 individuals in gnomAD carry two rare heterozygous missense variants, and that 2,689/44,514 have evidence of being in trans, making this a more frequent event. Here, higher levels of co-occurrence in the reference population indicate a higher probability that your observation in the proband has occurred by chance, cautioning against the overinterpretation of an observation of this nature in a patient without further evidence of pathogenicity.
The complete download table for all genes in the gnomAD v2 browser, encompassing five allele frequency thresholds (range ≤ 0.5% - ≤ 5%) and 26 functional variant consequence combinations can be found here.
Caveats and limitations
The variants considered in this analysis are exceedingly rare in the general population. This creates limitations in inferring phase (see “Variant Co-Occurrence (Phasing) Information in gnomAD”) and results in low overall counts of co-occurring damaging variants across the dataset. The absence of individuals with co-occurring variants in trans is therefore not an absolute indication that they cannot be tolerated by the gene and may result from an underpowered analysis needing a larger sample size. Our ability to identify rare variant pairs in trans in gnomAD v2 is also limited by the fact this dataset was used to calculate the Ptrans predictions. We find that our ability to detect in trans variant pairs therefore only extends down to an allele frequency of 0.5%, with all rarer combinations being dominated by unphased predictions and very few predictions of in trans variant pairs. The per-gene variant co-occurrence resource developed and released here is to be considered a first step in this space. We plan to use the Ptrans predictions from this dataset (gnomAD v2) on newer versions of gnomAD with additional samples in the future, where we can more confidently predict rare, in trans variant pairs, below an allele frequency threshold of ≤ 0.5%.
Conclusion
In summary, we leverage the size of gnomAD to provide a new resource to the medical genetics community, tabulating the co-occurrence of any two rare variants gene-wise across allele frequency and predicted functional variant consequences in the population. We hope that the information now displayed in the gnomAD v2 browser will aid clinicians and researchers in the interpretation of co-occurring variants. We encourage users to write to gnomad@broadinstitute.org with comments and suggestions for improving this feature.
Related post: Variant Co-Occurrence (Phasing) Information in gnomAD
- The highest allele frequency information for the non-bottlenecked populations—excluding Ashkenazi Jewish and European Finnish populations as well as individuals without an assigned population↩