Today, we are thrilled to announce the release of genome-wide structural variants (SVs) for 63,046 unrelated samples with genome sequencing (GS) data. All site-level information for 1,199,117 high-quality SVs discovered in these samples is browsable in the gnomAD browser (gnomAD SV v4) and downloadable from the gnomAD downloads page. For the full details of the v4 GS cohort (previously the v3 cohort), please refer to the prior blog posts regarding short variants (SNVs/indels) in gnomAD. These data represent the first SV dataset released as part of gnomAD that is native to the GRCh38 reference genome and should supersede the previous (hg19-based) gnomAD v2 SV callset for all GRCh38-based applications.
SVs in gnomAD v4 vs. v2: What’s new?
Before diving into the technical specifics of the GS-based SVs in gnomAD v4, we highlight some of the biggest improvements over the prior SV callset in gnomAD v2, including:

- Significantly greater scale: a total of 63,046 samples were jointly processed for SV discovery in v4 to produce ~1.2M SVs.
- Increased representation of SVs across a greater diversity of populations than gnomAD v2 (Figure 1).
- Updated definitions and annotations of complex SVs resolved by GATK-SV.
- Updated reference genome: all genomes in v4 were aligned to GRCh38 instead of hg19.
SV discovery methods
The new SV dataset for gnomAD v4 was generated using a cloud-based SV joint calling pipeline, GATK-SV (see Collins et al. 2020, PMID 32461652 for methodological details). GATK-SV is an ensemble approach that integrates results from multiple existing SV detection algorithms to maximize sensitivity while re-evaluating sequencing evidence directly to improve precision. In brief, SV calls from existing algorithms are merged and initially filtered using a random forest model that evaluates SVs with respect to alignment signatures including discordant paired-ends (PE), split reads (SR), read depth (RD), and B-allele frequencies (BAF). SVs are then jointly genotyped by integrating these signatures to assign genotypes and quality scores for each sample, and complex SVs (cxSVs) are resolved using a pattern matching approach. GATK-SV is publicly avalable at https://github.com/broadinstitute/gatk-sv. Specific to this project, we have greatly increased the scalability and improved the efficiency, sensitivity, and accuracy of GATK-SV to enable large-scale SV detection, yielding a median of 11,844 SVs per genome in gnomAD v4. The full technical details of these updated SV methods will be described in detail in an upcoming gnomAD v4 SV paper.
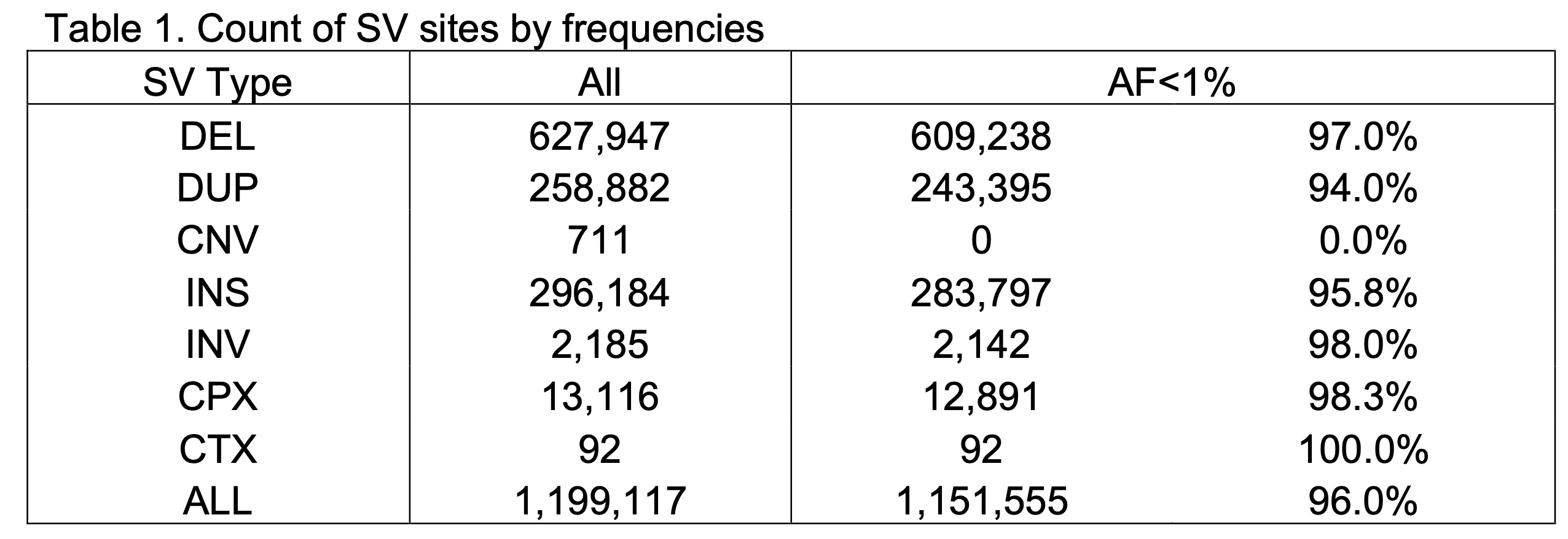
Description of SV sites
 and SVs per genome (B)")
The v4 SV callset represents 1,199,117 high-quality variant sites that have fully resolved alternate allele structures (Figure 2A, Table 1), including deletions (DEL), tandem duplications (DUP), insertions (INS), inversions (INV), multiallelic copy number variants (mCNV), reciprocal translocations (CTX), and complex rearrangements (CPX) (Figure 2A). An average of 11,844 SVs were detected in the individual genome across all SV types (Figure 2B). Consistent with our prior analyses in gnomAD v2 and other studies such as the 1000 Genomes Project (Byrska-Bishop et al. 2022, PMID 36055201), SVs in the human genomes are predominantly small and rare: the median SV size in gnomAD-SV v4 is 306 bp, and 96.0% of SVs in gnomAD v4 appear at allele frequencies (AF) below 1% in the population (Table 1).
Complex SVs and translocations
We have identified and resolved a total of 13,116 high-quality complex SV (CPX) sites that involve either two or more breakpoints or canonical SV signatures that cannot be explained by a single end-joining event. Approximately two-thirds of all events involve inversions (69.6%) while the remaining third are composed of various subtypes of dispersed duplications or insertions. We have also manually identified and resolved 92 canonical reciprocal translocations (CTX) in the 63,046 unrelated individuals of this callset, resulting in an estimated frequency of approximately 1.5 CTX per 1,000 individuals discovered at sequencing resolution.
Benchmarking SVs in gnomAD v4 against matched long-read sequencing data
, the Centers for Common Disease Genomics (CCDG), and the Trans-Omics for Precision Medicine (TOPMed) Program. (B) Precision of gnomAD v4 SV callset benchmarked by orthogonal long-read genome sequencing.")
We extensively evaluated the quality of the gnomAD v4 SV call set. Discovery of of SV counts identified in individual genomes in gnomAD v4 were compared against contemporary studies to estimate sensitivity (Figure 3A), and direct evaluation of SVs discovered from long-read genome sequencing data for 15 samples from the Human Genome Structural Variation Consortium (HGSVC) was used to evaluate precision (Figure 3B). For the long-read evaluation, we used two methods: (1) directly validating support for SVs in the long-read BAM file using VaPoR (Zhao et al., Gigascience, 2017, PMID 28873962) and (2) comparing the short-read SV calls from this callset against the long-read SV calls generated by the Human Genome Structural Variant Consortium (HGSVC) (Ebert et al. Science, 2021, PMID 33632895). Based on these analyses, we estimated that 86.7% of all SVs in gnomAD v4 were supported by corresponding long-read data in the same samples. Overall, there was higher long-read support rates for deletions and insertions (90.0% and 96.2% respectively) than duplications (73.0%) and inversions (85.8%) - though notably SV discovery for the latter two variant classes are more challenging to validate from long-read datasets in repetitive sequences (see Ebert et al., Science, 2021, PMID 33632895 and Porubsky et al., Cell, 2022, PMID 35525246 for review). We have previously shown that the majority of the >25,000 SVs in each genome that can only be captured by long-read assemblies reside in the most repetitive 9.7% of the hg38 reference assembly primarily composed of segmental duplications (SD) and simple repeats (SR) (see Zhao et al., AJHG 2021, PMID 33789087 for specific regions). When excluding this 9.7% of SD and SR sequences to estimate the precision of SVs in gnomAD, the precision of the callset increased to 96.9%, with false discovery rates across all SV types of 2.8% for deletions, 7.3% for duplications, 3.7% for insertions, and 7.0% for inversions, Figure 3B). Importantly, 90.9% of all protein coding exons in hg38, and 95.9% of all genes present in OMIM, span this 90.3% of the genome that does not include SD and SR sequences.
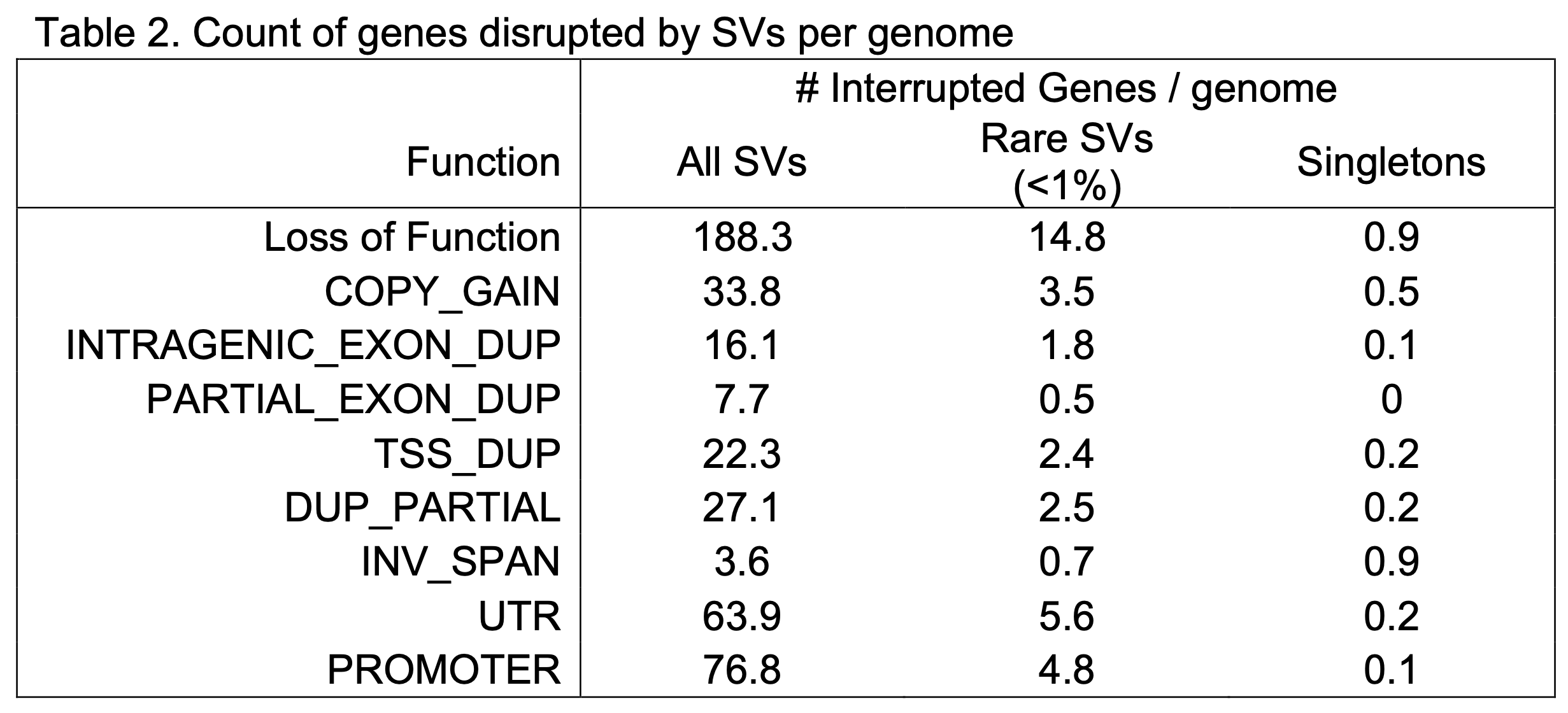
Functional impact
We have annotated the predicted functional impact of SVs using the GATK SVAnnotate tool with GENCODE release 39 (Figure 4). We found an average of 188 genes per genome in which the open reading frame was predicted to be altered by SVs and resulted in probable loss-of-function (pLoF), as well as 34 full gene duplications in each genome (Copy Gain [CG]). We have also annotated variants and genes with less clear functional impacts such as genes harboring nested duplications of one or more complete exons (Intragenic Exon Dup [IED]), or intragenic duplications for which at least one exon harbored a duplication breakpoint (Partial Exon Dup [PED]), duplications that include a transcription start site (TSS) but do not duplicate the entire gene (TSS Dup, TSSD), those that duplicate parts of a gene without the TSS (Dup Partial, DP), and genes inverted but fully intact (INV Span, IS). Counts of different functional classifications per person in v4 are displayed in Table 2.

Query SVs in gnomAD browser
Frequent users of the gnomAD browser will be glad to hear that the functionality for querying the gnomAD v4 SV callset has remained consistent with the gnomAD v2 SVs, which was originally described here:
https://gnomad.broadinstitute.org/news/2019-03-structural-variants-in-gnomad/.
Updated in January 2025 to fix Figure 1 image.