A critical component to the medical and functional interpretation of genetic variants involves the accurate estimation of their frequency. A variant’s frequency is defined by the number of copies of an allele divided by the total number of chromosomes examined.
The gnomAD database aggregates and harmonizes data across many large-scale sequencing projects to create summary allele frequency statistics. We stratify our aggregate frequencies by both sex and genetic ancestry group to aid in variant interpretation.
When and how to use genetic ancestry and population descriptors has been widely discussed, and we are keenly aware that there are limitations to using genetic ancestry groups. In this blog post, we will discuss what we mean by genetic ancestry, some imperfections of our current genetic ancestry inference methods, and why we feel group-stratified frequencies are critical to certain gnomAD applications that use gnomAD.
Genetic ancestry inference in gnomAD
Genetic ancestry is distinct from either race or ethnicity. Genetic ancestry, as defined by the National Academies of Sciences, Engineering, and Medicine (NASEM)1, reflects an individual’s demographic history and refers to the specific lines of descent through a family tree by which an individual inherited DNA from specific ancestors. Race and ethnicity, on the other hand, are sociopolitical constructs used to group individuals based on perceived shared ancestry or biological characteristics (race) or on perceived shared cultural heritage (ethnicity). Note that genetic ancestry is not equivalent to and does not negate how individuals self-identify.
Particularly given that genetic ancestry-based groupings have been misappropriated with malicious intent2, we think it is worth reiterating that assigning samples into discrete genetic ancestry-based groups does not accurately reflect our complex demographic history. Genetic ancestry is a continuous measure, so any methods of creating discrete groups of individuals will inherently be inadequate. Furthermore, we want to emphasize that any groupings of individuals, including the genetic ancestry groups annotated in gnomAD, are artificially created and are not naturally occurring.
In gnomAD, we use genetic similarity between samples to infer and create genetic ancestry groups. As described previously3,4,5, we perform a principal component analysis (PCA) on a set of high-quality SNVs to identify clusters of samples based on their genetic similarity, and these clusters roughly correspond to geographic ancestry provided by data contributors. We then train a random forest (RF) classifier on a subset of samples with provided genetic ancestry labels using the principal components from the PCA as features. Labels for each cluster have generally been based upon the International Genome Sample Resource, with some exceptions as described below. The gnomAD PCA loadings and RF classifier are both publicly available, and all of the underlying code is open-source.
Why gnomAD uses genetic ancestry groupings
Despite the known limitations of using genetic ancestry groups, we feel that stratification of our aggregate frequency information by group is still useful for the wider scientific community for two major reasons: variant classification and risk assessment.
Certain variants occur at elevated frequencies within specific genetic ancestry groups due to demographic history, such as bottleneck events. Being able to distinguish between the allele frequency across the entire gnomAD dataset (“global” AF) and within specific genetic ancestry groups (group maximum AF or grpmax) can help reduce the variant search space in disease diagnosis3. A variant may occur at high enough allele frequencies in a certain genetic ancestry group to rule out pathogenicity despite having a low global allele frequency. In addition, disease prevalences are often collected and reported with respect to specific geographical regions, and the ability to correlate genetic disease prevalence with observed disease prevalence can help inform when disease risk differs across genetic ancestry groups. We are aware that a few of our genetic ancestry groups (asj, ami, fin) should not be stratified at the same level as our other geography-based labels for ideological reasons. However, we have maintained these group labels to more easily identify variants that occur at different frequencies in different groups or regions of the world. For example, one of the most common variants, one of 39 Finnish Disease Heritage (FDH) diseases6, has an AF 50x greater in Finnish Europeans than other non-Finnish Europeans in gnomAD v2.
Sample metadata labels
We used the following genetic ancestry group labels to train our RF classifier for gnomAD v4:
- afr: African/African American
- ami: Amish
- amr: Admixed American
- asj: Ashkenazi Jewish
- eas: East Asian
- fin: Finnish
- mid: Middle Eastern
- nfe: European (non-Finnish)
- sas: South Asian
The genetic ancestry group labels were aggregated and harmonized using the following labels:
-
Harmonized labels from HGDP and 1KG: https://docs.google.com/spreadsheets/d/1jenSz_HnbA1kBESaUmur3Ob72-EPXJgfUWhbz5UdltA/edit#gid=0
-
Labels provided by different sequencing projects:
- African-American
- Amish
- Arab
- Ashkenazi Jewish
- British
- Bulgarian
- Canadian
- Continental African
- Costa Rican
- Dutch
- East Asian
- Estonian
- Finnish
- Finland
- German
- Hawaiian
- Hong Kong
- Italian
- Korean
- Native American
- Norwegian
- Pakistani
- Persian
- Singapore
- South Asian
- Spanish
- Swedish
- Sweden
- Taiwanese
- Tayside region of Scotland
- USA
Note that while some labels indicate a sample’s geographical origin (e.g., Finland), we do not know how all of these labels were collected or in what context.
Technical limitations of gnomAD genetic ancestry groups
As mentioned above, any methods that attempt to create discrete groups using genetic ancestry, which is a continuous measure, will be inherently inadequate. An additional limitation for genetic ancestry inference in gnomAD is inconsistency within provided sample metadata. The PCA used to cluster samples is label-agnostic, but the RF classifier relies on metadata labels given to us from various studies. The gnomAD project aggregates data and metadata; we do not directly recruit individuals or modify the provided metadata. This leads to large variability in metadata availability, specificity, and collection methodology, all of which hinder our ability to fully identify and define genetic ancestry groups. In addition, PCA is highly sensitive to the samples included in the dataset; samples from specific groups will not form distinct clusters unless there is adequate representation of that group within the dataset. This means that the PCs would be different and we might have more clusters if there were larger counts of samples from under-represented genetic ancestry groups. Within gnomAD v4, we are unable to label 31,256 samples, as they do not cluster with any of our reference metadata; these samples are included in our release in a group called “Remaining individuals”.
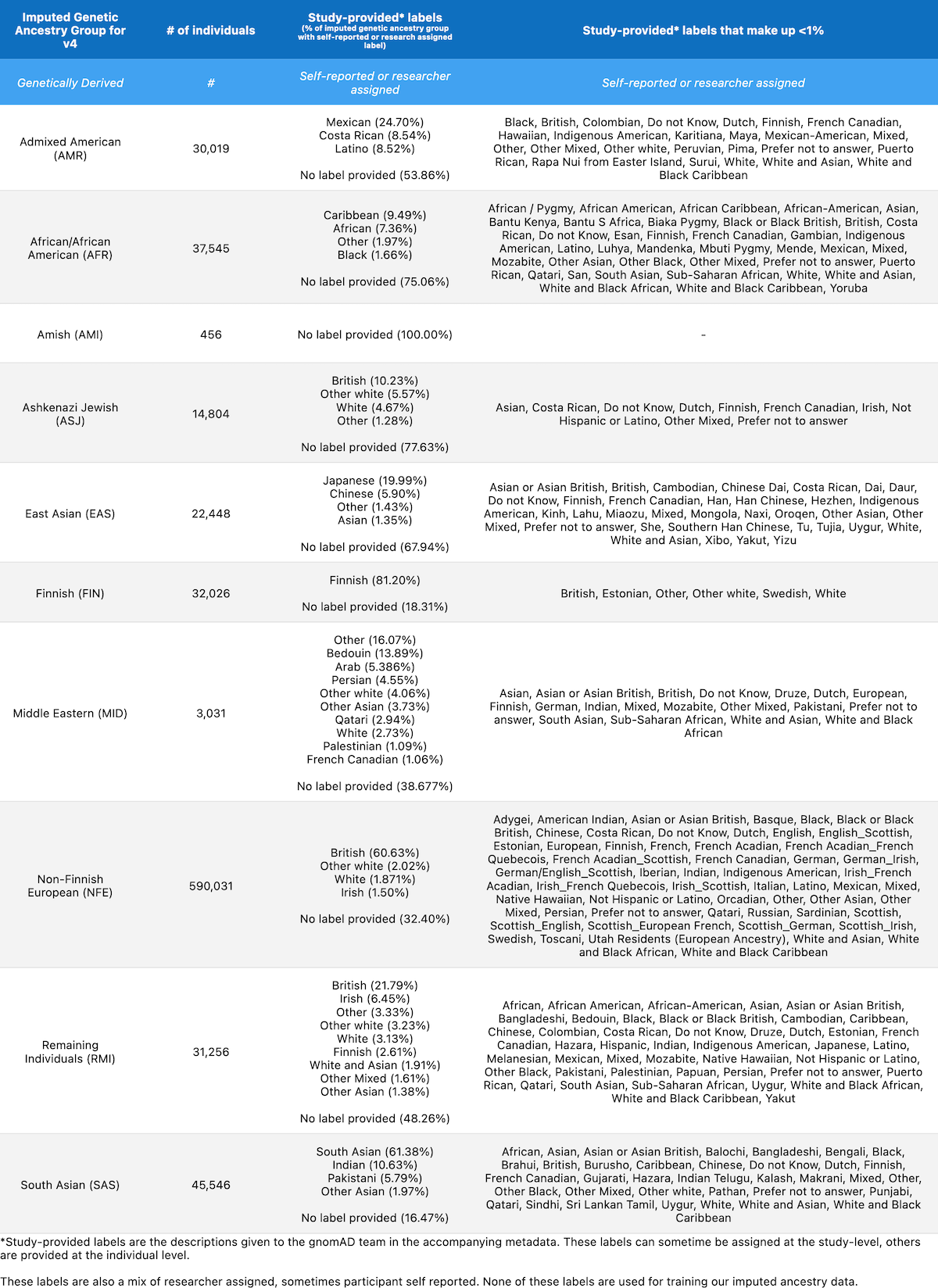
We share the following table in order to present how our inferred genetic ancestry groups correspond to descriptors provided by each contributing study. The table below lists the total number of individuals in each genetic ancestry group and the percentage of samples per group with each study-provided descriptor. Any descriptors that contribute to fewer than 1% of the samples per group have been aggregated into the column “Study-provided labels that make up less than 1%“. Note that the study-provided descriptors in the table below were not used to train our genetic inference model.
Continuing the conversation around genetic ancestry
All of the members of the gnomAD project continue to discuss best approaches to defining genetic ancestry across the gnomAD dataset. We are constantly refining the balance between accurately depicting the genetic ancestry of gnomAD samples and facilitating downstream use and application of our dataset. Our current method clusters genetically similar individuals, which leads to groupings that are imperfect representations of human history but which enable clinical variant classification.
A couple updates we have made in our current release are around terminology. We have updated all references of the term “population” within the browser to “genetic ancestry group” to indicate that these groups are inferred, artificially-created clusters of individuals. We have updated the term “Other” to “Remaining individuals” to reflect that we are unable to label these individuals with our current methods. And finally, we have changed “Latino/Admixed American” to “Admixed American” to avoid linking race and ethnicity with genetic ancestry inference.
Moving forward, we aim to tackle gnomAD content by increasing the diversity within gnomAD, particularly aiming to increase representation for under or non represented groups. This increase in representation will increase our understanding of the landscape of human genetic variation and will underscore the complex, continuous nature of genetic ancestry. In addition to enabling better science, increasing diversity in gnomAD datasets can help with reducing health inequities for groups that have been historically excluded and under-served7.
With adequate diverse representation, our method of inferring genetic ancestry will become obsolete. In the future, we plan to move away from genetic ancestry group-stratified frequencies and towards local or haplotype-based frequencies, and we plan to increase the granularity of our local ancestry inference where possible.
We welcome continued dialogue to ensure that gnomAD best serves the community and is respectful of scientists and study participants that have voluntarily shared their data. We encourage users to also consider and prevent potential misuse of their research findings, particularly with regard to genetic ancestry-based groupings. We point readers to an article entitled “Counter the weaponization of genetics research by extremists” that provides helpful guidance in this area.
To continue this conversation, or to provide any feedback or suggestions, contact us at gnomad@broadinstitute.org.
Acknowledgments
We would like to thank Alham Saadat in particular for her careful review of this blog post.
References
- https://www.nationalacademies.org/our-work/use-of-race-ethnicity-and-ancestry-as-population-descriptors-in-genomics-research
- Carlson, J., Henn, B. M., Al-Hindi, D. R., & Ramachandran, S. (2022). Counter the weaponization of genetics research by extremists. Nature, 610(7932), 444–447. https://doi.org/10.1038/d41586-022-03252-z
- Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E.*, Banks, E., Fennell, T., O’Donnell-Luria, A. H., Ware, J. S., Hill, A. J., Cummings, B. B., Tukiainen, T., Birnbaum, D. P., Kosmicki, J. A., Duncan, L. E., Estrada, K., Zhao, F., Zou, J., Pierce-Hoffman, E., … Daly, M. J., MacArthur, D. G. & Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). https://doi.org/10.1038/nature19057
- Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., Collins, R. L., Laricchia, K. M., Ganna, A., Birnbaum, D. P., Gauthier, L. D., Brand, H., Solomonson, M., Watts, N. A., Rhodes, D., Singer-Berk, M., England, E. M., Seaby, E. G., Kosmicki, J. A., … MacArthur, D. G. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). https://doi.org/10.1038/s41586-020-2308-7
- Chen, S., Francioli, L. C., Goodrich, J. K., Collins, R. L., Wang, Q., Alföldi, J., Watts, N. A., Vittal, C., Gauthier, L. D., Poterba, T., Wilson, M. W., Tarasova, Y., Phu, W., Yohannes, M. T., Koenig, Z., Farjoun, Y., Banks, E., Donnelly, S., Gabriel, S., Gupta, N., Ferriera, S., Tolonen, C., Novod, S., Bergelson, L., Roazen, D., Ruano-Rubio, V., Covarrubias, M., Llanwarne, C., Petrillo, N., Wade, G., Jeandet, T., Munshi, R., Tibbetts, K., gnomAD Project Consortium, O’Donnell-Luria, A., Solomonson, M., Seed, C., Martin, A. R., Talkowski, M. E., Rehm, H. L., Daly, M. J., Tiao, G., Neale, B. M.†, MacArthur, D. G.† & Karczewski, K. J. A genome-wide mutational constraint map quantified from variation in 76,156 human genomes. bioRxiv 2022.03.20.485034 (2022). https://doi.org/10.1101/2022.03.20.485034
- Uusimaa, J., Kettunen, J., Varilo, T., Järvelä, I., Kallijärvi, J., Kääriäinen, H., Laine, M., Lapatto, R., Myllynen, P., Niinikoski, H., Rahikkala, E., Suomalainen, A., Tikkanen, R., Tyynismaa, H., Vieira, P., Zarybnicky, T., Sipilä, P., Kuure, S., & Hinttala, R. (2022). The Finnish genetic heritage in 2022 - from diagnosis to translational research. Disease models & mechanisms, 15(10), dmm049490. https://doi.org/10.1242/dmm.049490
- Fatumo, S., Chikowore, T., Choudhury, A., Ayub, M., Martin, A. R., & Kuchenbaecker, K. (2022). A roadmap to increase diversity in genomic studies. Nature medicine, 28(2), 243–250. https://doi.org/10.1038/s41591-021-01672-4
- Lewis, A. C. F., Molina, S. J., Appelbaum, P. S., Dauda, B., Di Rienzo, A., Fuentes, A., Fullerton, S. M., Garrison, N. A., Ghosh, N., Hammonds, E. M., Jones, D. S., Kenny, E. E., Kraft, P., Lee, S. S., Mauro, M., Novembre, J., Panofsky, A., Sohail, M., Neale, B. M., & Allen, D. S. (2022). Getting genetic ancestry right for science and society. Science (New York, N.Y.), 376(6590), 250–252. https://doi.org/10.1126/science.abm7530
- Cardon, L. R., & Palmer, L. J. (2003). Population stratification and spurious allelic association. Lancet (London, England), 361(9357), 598–604. https://doi.org/10.1016/S0140-6736(03)12520-2
- Ziv, E., & Burchard, E. G. (2003). Human population structure and genetic association studies. Pharmacogenomics, 4(4), 431–441. https://doi.org/10.1517/phgs.4.4.431.22758
- Tiwari, H. K., Barnholtz-Sloan, J., Wineinger, N., Padilla, M. A., Vaughan, L. K., & Allison, D. B. (2008). Review and evaluation of methods correcting for population stratification with a focus on underlying statistical principles. Human heredity, 66(2), 67–86. https://doi.org/10.1159/000119107
- Mathieson, I., & Scally, A. (2020). What is ancestry?. PLoS genetics, 16(3), e1008624. https://doi.org/10.1371/journal.pgen.1008624