Today we are pleased to announce the incorporation of manual loss-of-function (LoF) curations into the gnomAD v2.1.1 browser. As of this release, we have curated all homozygous pLoFs and a small set of recessive genes (e.g., GAA, GLA, IDUA, SMPD1, GBA, FIG4, MCOLN1, AP4B1, AP4M1, AP4S1, and AP4E1). These curations were performed for multiple projects including the recently published work, Karczewski et al. 2020 Nature, as well as other gene-specific projects. We are so excited to start sharing this data with you that we are including it in the gnomAD v3.1 release announcement but really these are a new gnomAD v2.1.1 feature at the moment. More datasets will be added to the browser as they are completed.
What is loss-of-function curation?
The classification of LoF variants is a result of a specialized and manual curation of predicted loss-of-function (pLoF) variants that have passed all LOFTEE filters and other QC flags in gnomAD to determine how likely these variants are to result in loss of function. After reviewing all the high-quality homozygous pLoF variants in gnomAD, we determined that 28% may not actually result in loss-of-function, highlighting the need for careful curation of these variants. Curation results also correlated well with our biological expectation; for heterozygous variants, 60% of pLoF variants in a set of haploinsufficient disease genes—versus 25% in a set of recessive disease genes—did not appear to result in loss-of-function.
pLoF curated variants were assigned one of five classifications based on the presence or absence of certain error modes. These classifications include: LoF, likely LoF, uncertain LoF, likely not LoF, and not LoF. For each curated variant, two biocurators performed an independent curation, and discrepancies between biocurators were discussed and resolved as a team.
Variants classified as “not LoF” have error modes that indicate these variants are predicted to not result in loss of function, while “LoF” classified variants have no error modes (or at most minor error modes) that indicate they are predicted to result in loss-of-function. Similar to the use of the “likely” qualifier in the ACMG/AMP criteria for sequence variant interpretation for classifying likely pathogenic and likely benign variants, “likely LoF” and “likely not LoF” classified variants are slightly less confidently predicted to result in loss-of-function or not loss-of-function, respectively. Variants with an “uncertain LoF” classification are similar to the ACMG/AMP variants of uncertain significance (VUS’s), and do not have sufficient evidence to point towards a definitive classification. We recommend using these classifications in coordination with applying PVS1 in the ACMG/AMP criteria for sequence variant interpretation.
How to use these annotations
For “likely not LoF” and “not LoF” variants, our extensive manual curation supports the conclusion that these variants may either be the result of a technical sequencing error, or their predicted biological effect is not loss-of-function. For those variants with technical errors that are also deemed “not LoF,” the allele frequency of these variants in gnomAD should not be considered the true frequency. This means that if you observe (and have confirmed) the variant in a patient, you should not use the gnomAD allele frequencies to estimate the prevalence of this variant — particularly for cases where there is a flag suggesting that the variant in gnomAD appears to be an artifact in at least some of the individuals.
For “likely LoF” and “LoF” variants, our manual curation provides increased confidence that these variants are truly loss-of-function variants and are expected to result in nonsense-mediated decay or an otherwise null effect. This systematically excludes a number of common annotation and sequencing error modes. Ultimately, however, functional studies will be needed to fully validate the potential loss-of-function impact of a variant.
“Uncertain LoF” variants represent cases where we were unable to reach a more conclusive classification and therefore should not be interpreted as falling into either of the above two categories.
Loss-of-function curations in the gnomAD browser
These curations are viewable from both the gene and variant pages on the browser. Within the gene page there is a new column that lists the curation result for all variants for which curation is available. If you hover over the curation result, you will see the error flag(s) that were identified during the curation of that variant. To date, curation has been performed on over 4,000 high-confidence pLoF variants in the gnomAD dataset, but this is a small fraction of the 444,000 total pLoF variants. As additional curations are performed, we will continue to update the gnomAD browser.
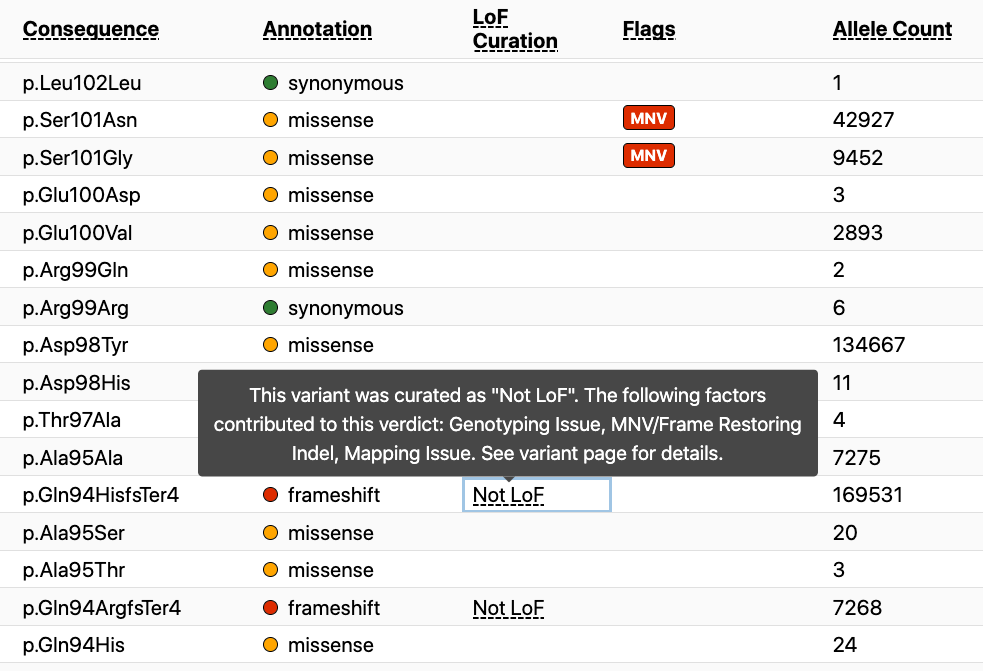
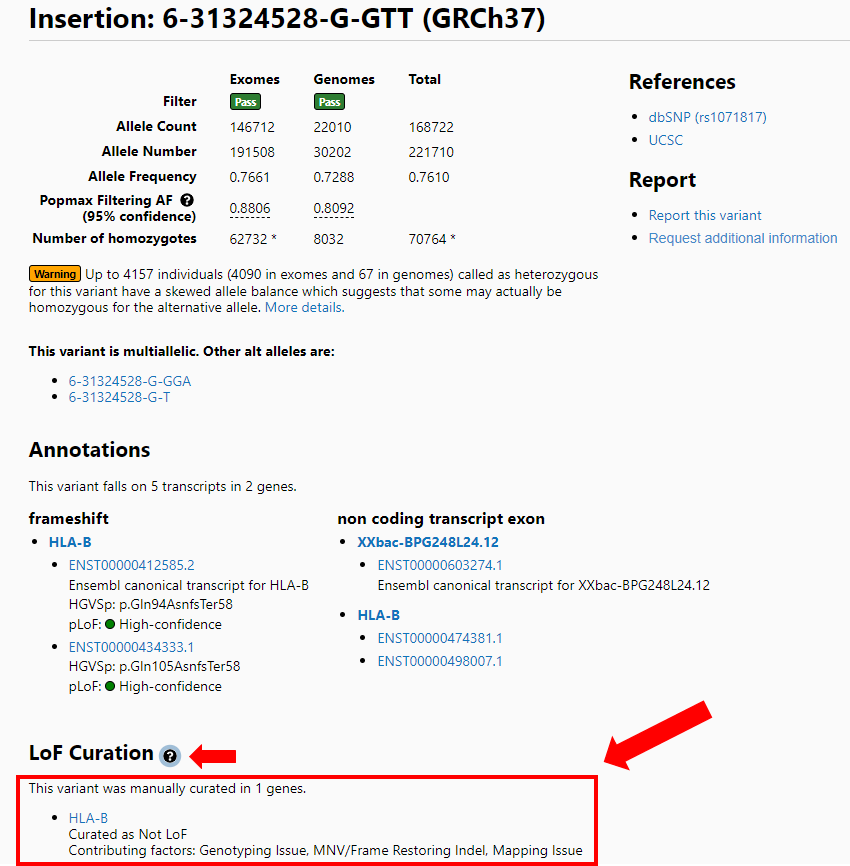
On the variant page there is an LoF curation section that also lists the flag(s) present during the curation of this variant with the verdict it received as a result of the curation (large red arrow). A popup (small red arrow) is available for a detailed explanation of these error modes from the variant page as well.
Caveats
These curations are based on the presence of these variants in the gnomAD database and do not take into account the presence of the variant in individuals with documented disease. We view LoF curation as an important step to be taken to evaluate whether a variant is expected to result in loss-of-function and would follow this with curation for the clinical impact of a variant (e.g., pathogenic, benign, etc.) following the ACMG/AMP standards and guidelines for the interpretation of sequence variants (Richards et al. 2015), including whether loss-of-function is a mechanism of disease for the specific gene. Therefore, even if a variant is classified as “LoF” or “likely LoF,” it may not meet criteria for pathogenicity with respect to human disease.
Additionally, the gnomAD LoF curation of a variant is not in any way determined based on functional evidence. Rather, predictions are entirely based on manual interpretation of splicing, conservation, computational annotations, and sequence data quality in gnomAD and the UCSC genome browser.
Acknowledgements
We would like to thank all the people who have been critical in the completion of these projects. This includes an amazing group of biocurators, especially Eleanor Seaby, Eleina England, Dan Rhodes, Rachel Son, and Emily Evangelista. In addition, the curations could not have been completed without the support of software engineers Nick Watts, Ben Weisburd, and Matthew Solomonson. All of these projects were done in collaboration with gnomAD team members, including Konrad Karczewski, Beryl Cummings, Miriam Udler, and Julia Goodrich. Lastly, we want to acknowledge the support and guidance of this work by Samantha Baxter, Daniel MacArthur, and Heidi Rehm.